
Artigos recentes
Structural and electronic properties of amorphous silicon and germanium monolayers and nanotubes: A DFT investigation
Raphael M. Tromer, Marcelo L. Pereira Júnior , Luiz. A. Ribeiro Júnior , Douglas S. Galvão
A recent breakthrough has been achieved by synthesizing monolayer amorphous carbon (MAC). Here, we used ab initio (DFT) molecular dynamics simulations to study silicon and germanium MAC analogs. Typical unit cells contain more than 600 atoms. We also considered their corresponding nanotube structures. The cohesion energy values for MASi and MAGe are approximately 3.0 eV/atom lower than the energy ordering of silicene and germanene, respectively. Their electronic behavior varies from metallic to small band gap semiconductors. Since silicene, germanene, and MAC have already been experimentally realized, the corresponding MAC-like versions we propose are within our present synthetic capabilities.
Irida-graphene phonon thermal transport via non-equilibrium molecular dynamics simulations
Isaac M. Felix, Raphael M. Tromer, Leonardo D. Machado, Douglas S. Galvão, Luiz A. Ribeiro, Jr and Marcelo L. Pereira, Jr
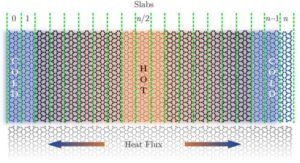
Recently, a new 2D carbon allotrope called Irida-Graphene (Irida-G) was proposed, and its reliable stability has been previously predicted. Irida-G is a flat sheet topologically arranged into 3-6-8 carbon rings exhibiting metallic and non-magnetic properties. In this study, we investigated the thermal transport properties of Irida-G using classical reactive molecular dynamics simulations. The findings indicate that IridaG has an intrinsic thermal conductivity of approximately 215 W mK−1 at room temperature, significantly lower than that of pristine graphene. This decrease is due to characteristic phonon scattering within IridaG’s porous structure. Additionally, the phonon group velocities and vibrational density of states for Irida-G were analyzed, revealing reduced average phonon group velocities compared to graphene. The thermal conductivity of Irida-G is isotropic and shows significant size effects, transitioning from ballistic to diffusive heat transport regimes as the system length increases. These results suggest that while Irida-G has lower thermal conductivity than graphene, it still holds potential for specific thermal management applications, sharing characteristics with other two-dimensional materials.
TH-graphyne: a new porous bidimensional carbon allotrope
Kleuton A. L. Lima, Rodrigo A. F. Alves, Daniel A. da Silva, Fábio L. L. Mendonça, Marcelo L. Pereira, Jr and Luiz A. Ribeiro, Jr
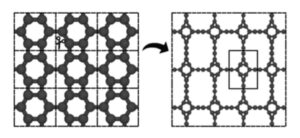
Graphyne and two-dimensional porous carbon-based materials have garnered significant attention due to their interesting structural characteristics and essential properties for new technological applications. Within this scope, this work investigates the structural, thermal, electronic, optical, and mechanical properties of a novel two-dimensional allotrope that combines triangular (T) and hexagonal (H) rings, connected by acetylenic linkages (graphyne-like), thus named TH-graphyne (TH-GY). This study comprehensively characterizes the proposed system’s behavior using density functional theory, ab initio molecular dynamics, and classical reactive molecular dynamics simulations. Our results confirm the structural stability of TH-GY. AIMD simulations demonstrate the material’s thermal stability at elevated temperatures, while phonon dispersions indicate its dynamical stability. Electronic band structure calculations show that the system is metallic. The analysis of optical properties reveals intense activity in the visible and UV regions, with pronounced anisotropy. A machine learning interatomic potentials model was developed for TH-GY and used to determine the mechanical behavior of the system, which exhibits Young’s modulus ranging from 263 to 356 GPa, highlighting its flexibility. Classical reactive MD simulations elucidate the fracture behavior of TH-GY, revealing distinct fracture patterns and mechanical anisotropy.
Can 2D Carbon Allotropes Be Used as Photovoltaic Absorbers in Solar Harvesting Devices?
Alexandre Cavalheiro Dias, Carlos Derli Almeida Cornélio, Maurício Jeomar Piotrowski, Luiz Antônio Ribeiro Júnior, Carlos Maciel de Oliveira Bastos, Celso Ricardo Caldeira Rêgo and Diego Guedes-Sobrinho
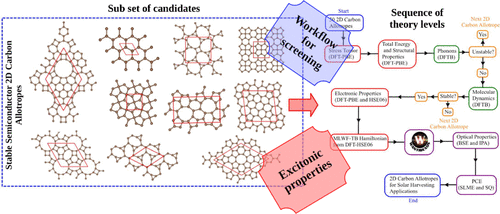
Sustainable energy solutions have led to extensive research into materials for the conversion of solar energy. Two-dimensional carbon allotropes have garnered significant attention due to their unique structural and electronic properties, which can enhance the efficiency and sustainability of solar panels. This study used several computational methods, including density functional theory, density functional tight binding, and molecular dynamics simulations, to explore the solar energy conversion capabilities of 30 different 2D carbon-based allotropes. After a thorough analysis, we found that these materials exhibited a wide range of power conversion efficiency values, from 7% to 30%, assuming complete absorption of incident light. Our research provides valuable insights into the structural and electronic properties that impact the performance of these materials in solar cell applications.
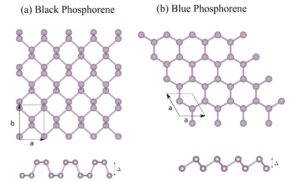
Signature of excitonic insulators in phosphorene nanoribbons
Andre Felipe Pereira de Oliveira, Andréia Luisa da Rosa and Alexandre Cavalheiro Dias
Phosphorene is a recently developed two-dimensional (2D) material that has attracted tremendous attention because of its unique anisotropic optical properties and quasi-one-dimensional (1D) excitons. We use first-principles calculations combined with the maximally localized Wannier function tight binding Hamiltonian and Bethe–Salpeter equation (BSE) formalism to investigate quasiparticle effects of 2D and quasi-1D blue and black phosphorene nanoribbons. Our electronic structure calculations shows that both blue and black monolayered phases are semiconductors. On the other hand black phosphorene zigzag nanoribbons are metallic. Similar behavior is found for very thin blue phosphorene zig-zag and armchair nanoribbon. As a general behavior, the exciton binding energy decreases as the ribbon width increases, which highlights the importance of quantum confinement effects. The solution of the BSE shows that the blue phosphorene monolayer has an exciton binding energy four times higher than that of the black phosphorene counterpart. Furthermore, both monolayers show a different linear optical response with respect to light polarization, as black phosphorene is highly anisotropic. We find a similar, but less pronounced, optical anisotropy for blue phosphorene monolayer, caused exclusively by the quasi-particle effects. Finally, we show that some of the investigated nanoribbons show a spin-triplet excitonic insulator behavior, thus revealing exciting features of these nanoribbons and therefore provides important advances in the understanding of quasi-one dimensional phosphorus-based materials.
Stability and Optoelectronic Properties of Two-Dimensional Gallium Phosphide
Elisangela da Silva Barboza, Kessia L. M. Cruz, Ramon S. Ferreira, Alexandre C. Dias, Erika N. Lima, Diego R. da Costa and Teldo A. S. Pereira
Using first-principles calculations, density functional theory, and the tight-binding method, we investigate the optoelectronic properties of two-dimensional gallium phosphide (2D GaP). Our investigation covers electronic properties, such as band structure and electronic band gap, and optical properties, including absorption spectra, refractive index, and reflectivity, considering excitonic effects. Additionally, structural aspects such as stability, elastic properties, and Raman and infrared spectra are also analyzed. This comprehensive study brings up valuable insights into 2D GaP physics, evincing the key features that make it a potential material for optoelectronic applications, such as photodetectors and solar cells.
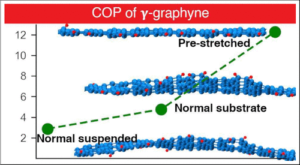
Enhanced Elastocaloric Effects in γ-Graphyne
Guilherme B. Kanegae, Marcelo L. Pereira Junior, Douglas S. Galvão, Luiz A. Ribeiro Junior and Alexandre F. Fonseca
The global emphasis on sustainable technologies has become a paramount concern for nations worldwide. Specifically, numerous sustainable methods are being explored as promising alternatives to the well-established vapor-compression technologies in cooling and heating devices. One such avenue gaining traction within the scientific community is the elastocaloric (eC) effect. This phenomenon holds promise for efficient cooling and heating processes without causing environmental harm. Studies carried out at the nanoscale have demonstrated the efficiency of the eC effect, proving to be comparable to that of state-of-the-art macroscopic systems. In this study, we used classical molecular dynamics simulations to investigate the elastocaloric effect for the recently synthesized γ-graphyne. Our analysis goes beyond obtaining changes in eC temperature and the coefficient of performance (COP) for two species of γ-graphyne nanoribbons (armchair and zigzag). We also explore their dependence on various conditions, including whether they are deposited on a substrate or prestrained. Our findings reveal a substantial enhancement in the elastocaloric effect for γ-graphyne nanoribbons when subjected to prestrain, amplifying it by at least 1 order of magnitude. Under certain conditions, the changes in the eC temperature and the COP of the structures reach expressive values as high as 224 K and 14, respectively. We discuss the implications of these results by examining the shape and behavior of the carbon–carbon bond lengths within the structures.
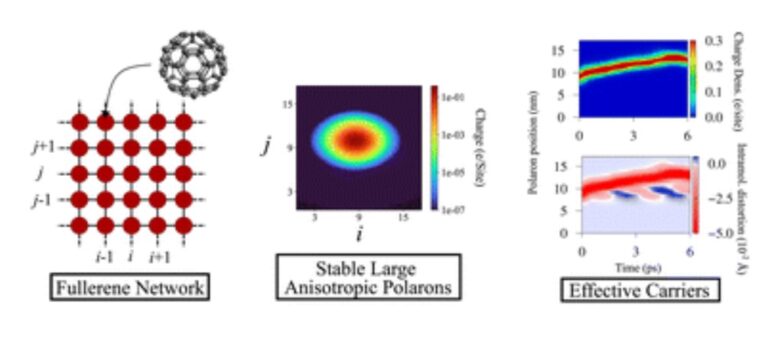
Large polarons in two-dimensional fullerene networks: the crucial role of anisotropy in charge transport
T. S. A. Cassiano, M. L. Pereira, Junior, G. M. e Silva, P. H. de Oliveira Neto and L. A. Ribeiro, Junior
The recent synthesis of a two-dimensional quasi-hexagonal-phase monolayer network of C60 molecules, known as qHPC60, holds significant promise for future semiconductor applications. However, the mechanism behind charge transport in these networks remains unknown. In this study, we developed a Holstein–Peierls Hamiltonian model to investigate charge transport in qHPC60, incorporating both local and non-local electron–phonon couplings. Our computational approach involved identifying suitable semi-empirical parameters to realize the formation of stable polarons in this material. The results unveiled the formation of stable large polarons as the primary carriers in the charge transport throughout qHPC60. To explore polaron transport properties, we conducted dynamic simulations within the picosecond time scale while subjecting the system to an external electric field. Our analysis emphasized the substantial influence of anisotropy on shaping mobile polarons, with an anisotropy coefficient of at least 50%. The polarons exhibited velocities within the acoustic regime ranging from 0.5–1.5 nm ps−1. While these velocities are comparable to those observed in high-end organic molecular crystals, they are considerably lower than those in graphene and conducting polymers. With qHPC60 possessing a semiconducting band gap of approximately 1.6 eV, our findings shed light on its potential application in flat electronics, overcoming the null-gap predicament of graphene.